Data Analysis Notebook - How to bring in data from a Gen3 Data Commons to the workspace and perform data analysis¶
1. Introduction to the Open Access Data Commons¶
- The Open Access Data Commons https://gen3.datacommons.io/ supports the management, analysis and sharing of data for the research community with the aim of accelerating discovery and development of diagnostics, treatment and prevention of diseases.
- Gen3 Data Commons store a) data files and b) structured metadata.
- For the first part of this notebook (sections 2 and 3), we show how to download data files and bring them to the workspace using the Gen3-client and in the second part below (section 4), we will show how to download structured metadata to the workspace using the Gen3 Python SDK.
2. Download data files from the Gen3 Data Commons and bring them to the workspace¶
2.1 Introduction to the dataset¶
- We will analyze two data files ('GSE63878_final_list_of_normalized_data.txt.gz' and 'pheno_63878_2.txt') from the study "GEO-GSE63878".
- This study deals with peripheral blood leukocytes gene expressions which were subject to transcriptional analysis for 48 service members both prior-to and following deployment to conflict zones. Half of the subjects returned with Post-traumatic Stress Disorder (PTSD), while the other half did not.
2.2 Importing the data files to the workspace using the Gen3-client: a step-by-step guide¶
- First, we can find and browse all data files stored on the Gen3 Data Commons under the "Files" tab on the Data Exploration page.
To download data files, we will create and download a file manifest, which is a light JSON file that is called by the Gen3-client to download all enlisted entities to the workspace:
In the Explorer under the "Files" tab we find the "Data Format" category; from here we can select the box next to "TXT" that builds a cohort and shows all files in the Data Commons that end on "TXT". In this case: 'GSE63878_final_list_of_normalized_data.txt.gz' and 'pheno_63878_2.txt'.
- We click on "Download (File) Manifest", save it to our local drive, and upload it to the workspace under the /pd directory as "file-manifest.json". For help on this step, see the screen recordings shown here.
- Only the files in the /pd directory will persist in the cloud after workspace termination.
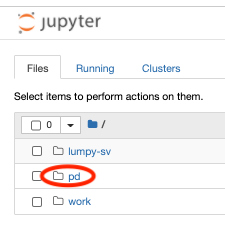
- We visit now the profile page, click on "Create API key", download the .JSON file and upload this "credentials.json" to the workspace under the /pd directory.
- In the workspace, we open a new terminal.
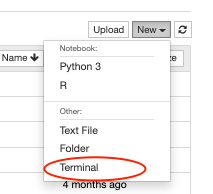
- We run the following commands in the terminal (also shown here) to download and install the Gen3-client, configure the profile "demo" with the "credentials.json", and to download the data files calling the "file-manifest.json":
- wget https://github.com/uc-cdis/cdis-data-client/releases/download/2020.11/dataclient_linux.zip
- unzip dataclient_linux.zip
- PATH=$PATH:~/
- gen3-client configure --apiendpoint=https://gen3.datacommons.io --profile=demo --cred=~/pd/credentials.json
- cd pd
- gen3-client download-multiple --profile=demo --manifest=file-manifest.json --skip-completed- The two files should be now saved in the /pd directory. You can terminate the terminal session.
Note. If you want to download only a single data file the Gen3-client command changes as shown here. You can also find the data file on the Exploration Page and click on the file's GUID to "Download".
3. Load and analyze the data files here in the workspace¶
- For this section, you need to start running a jupyter python notebook and run the code snippets below.
3.1 Install dependencies and import python libraries¶
# Uncomment the lines to install libraries if needed.
# !pip install numpy
# !pip install matplotlib
# !pip install pandas
# !pip install seaborn
# Import libraries:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import matplotlib as mpl
import os
import seaborn as sns
import re
from pandas import DataFrame
import warnings
warnings.filterwarnings("ignore")
import gzip
import scipy
import sys
import sklearn
import random
import math
3.2 Unzip data file¶
!gzip -dk 'GSE63878_final_list_of_normalized_data.txt.gz' # command -k saves the original zipped file
3.3 Load the first txt file as a Pandas dataframe "pheno_df"¶
This dataframe shows the characteristics, sample descriptions, etc. associated with measured gene expression.
pheno_df = pd.read_csv('/home/jovyan/pd/pheno_63878_2.txt', sep='\t')
pheno_df.head() # show top 5 rows of dataframe
3.4 Load the second txt file as a Pandas dataframe "rna_df"¶
This dataframe shows genome expressions. Numbers after "Sample_" indicate pre-deployment ("1") and post-deployment ("3").
os.chdir('/home/jovyan/pd')
rna_df = pd.read_csv('/home/jovyan/pd/GSE63878_final_list_of_normalized_data.txt', sep='\t')
rna_df.head()
3.5 Prepare the second dataframe rna_df (e.g. data cleaning)¶
rna_df = rna_df.dropna(1) # remove columns that contain "NaN"
del rna_df['Probe ID'] # delete first column for further analysis.
all_genes = set(rna_df["Gene Symbol"].to_list()) # save this column as list for further analysis.
3.6 Organize pheno_df and rna_df data into categories and combine¶
# list(pheno_df.columns)
trim_pheno_df = pheno_df[['Comment [Sample_description]', 'Characteristics [condition]', 'FactorValue [time-point]']] # select columns to be worked with
trim_pheno_df.head()
# Add the categories to the dataset
blank = [name for name in rna_df.columns] # list all headers in rna_df
# Category "condition"
condition = (trim_pheno_df['Characteristics [condition]']).tolist() # move all rows of this column into a list
condition = condition[::-1] # switch as column Characteristics [condition] begins with sample48_3 instead of Sample1_1
condition.insert(0, 'condition') # add header
ptsd = {col:val for col, val in zip(blank, condition)} # match headers from rna_df to 'condition'
# Category "deployment"
deployment = (trim_pheno_df['FactorValue [time-point]']).tolist()
deployment = deployment[::-1]
deployment.insert(0, 'deployment')
deploy = {col:val for col, val in zip(blank, deployment)}
Attention: **The next two code snippets should be run only once.**
# Adding category lists to the rna_df dataframe
# This will combine both datasets
rna_df = rna_df.append(ptsd, ignore_index=True) # run only once
rna_df = rna_df.append(deploy, ignore_index=True) # run only once
rna_df.tail() # shows the last 5 rows of the dataframe
# Transpose and relabel for easy wrangling
trans = rna_df.transpose()
trans.columns = trans.iloc[0] # [0] is the gene symbol row
trans = trans.drop(trans.index[0]) # only run once, or you'll start losing genes
trans.head()
3.7 Statistical analysis on data¶
- First we define the analysis functions and then we plot the data.
3.7.1 Processing dataframe¶
# Import libraries
import scipy
import sys
import sklearn
import random
import math
# Define function
def process_data(expression_df, condition, control, experimental):
#expresion_df = input is the dataframe that we have defined above, gene expressions before and after deployment
#condition = choose condition; for example separate your dataframe between between condition and deployment; input as string
#control = control variable; input as string
#experimental = experimental variable, input as string
#returns dataframe of gene names, mean values, log2fold change, p-value, -log10(pval), and all replicates for each gene
experimental_df = expression_df[expression_df[condition].str.contains(experimental)]
experimental_df = expression_df.drop(columns=['condition', 'deployment'])
control_df = expression_df[expression_df[condition].str.contains(control)]
control_df = control_df.drop(columns=['condition', 'deployment'])
deg_genes = {} # dictionary of final data
gene_names = list(experimental_df.columns)
for gene in gene_names:
ex_mean = experimental_df[gene].mean() # experimental mean
ctrl_mean = control_df[gene].mean() # control mean
ex_reps = experimental_df[gene] # all replicates of PTSD samples
control_reps = control_df[gene] # all replicates of control samples
pval = scipy.stats.ttest_ind(control_reps, ex_reps) # calculate pval
pvalue = pval.pvalue # gets specific p-value, removes meta data
gene_data = {
'GeneNames': gene,
'ctrl_mean': ctrl_mean,
'ex_mean': ex_mean,
'log2(foldchange)': math.log2(ex_mean) - math.log2(ctrl_mean),
'p-value': pvalue, #gets only the p-val
'-log10(p-value)': math.log10(pvalue) * (-1),
'ctrl_reps': control_reps.values.tolist(),
'experimental_reps': ex_reps.values.tolist()
}
deg_genes[gene] = gene_data
deg_data_frame = pd.DataFrame.from_dict(deg_genes, orient='index')
return(deg_data_frame)
# Returns dataframe of gene names, means, log2fold change, p-value, -log10(pval), and all replicates for each gene
deg_data_frame = process_data(trans, 'condition', 'control', 'PTSD')
deg_data_frame.reset_index(drop=True)
3.7.2 Plot top gene expressions¶
# Define function
def top_expressed_gene(deg_data_frame, control, experimental, top_number):
# requires deg_data_frame from process data, string of control and experimental mean names, and top number of genes
# returns plot of top expressed genes in the experimental group, plotted against the expression of the control group
control_mean = deg_data_frame[control]
experimental_mean = deg_data_frame[experimental]
sorted_mean = experimental_mean.sort_values(ascending= False) # sorting by greatest expression
top_genes = sorted_mean[:top_number].keys().tolist() # getting the top expressed genes
control_vals = deg_data_frame['ctrl_reps'][top_genes]
experimental_vals = deg_data_frame['experimental_reps'][top_genes]
expression_data = pd.DataFrame([control_vals, experimental_vals])
print('The top ' +str(top_number)+ ' expressed genes are:' )
for gene in top_genes:
sns.set(style='whitegrid')
plot_data = expression_data[gene].apply(pd.Series)
new_plot_data=plot_data.T
new_plot_data.columns =['Control', 'Experiment']
sns.violinplot(data=new_plot_data, palette="Set1").set(title=str(gene))
ax = sns.swarmplot(data=new_plot_data, color="0", alpha=.35)
ax.set(ylabel='Expression')
plt.show()
# Returns plot of top expressed genes in the experimental group, plotted against the expression of the control group
top = top_expressed_gene(deg_data_frame, 'ctrl_mean', 'ex_mean', 2)


3.7.3 Plot favorite gene expression¶
# Define function
def your_fav_gene(deg_data_frame, control, experimental, fav_gene):
# requires deg_data_frame from process data, string of control and experimental names, and name of gene you'd like to plot
# returns plot of expression in control and experimental group
control_mean = deg_data_frame[control][fav_gene]
experimental_mean = deg_data_frame[experimental][fav_gene]
control_vals = deg_data_frame['ctrl_reps'][fav_gene]
experimental_vals = deg_data_frame['experimental_reps'][fav_gene]
expression_data = pd.DataFrame([control_vals, experimental_vals])
#print('Favorite expressed gene: ' +str(fav_gene))
#print(expression_data)
sns.set(style='whitegrid')
plot_data = expression_data.transpose()
plot_data.rename(columns = {0:'Control',1:'Experiment'}, inplace=True)
ax = sns.violinplot(data=plot_data, palette="husl").set(title='Your favorite gene is '+str(fav_gene))
ax = sns.swarmplot(data=plot_data, color="1", alpha=.4)
ax.set(ylabel='Expression')
plt.show()
# Returns plot of expression in control and experimental group of the gene of our choice
ELMO2 = your_fav_gene(deg_data_frame, 'ctrl_mean', 'ex_mean', 'ELMO2') # change to any gene in 'ELMO2'

# Returns plot of expression in control and experimental group of the gene of our choice
ZNHIT1 = your_fav_gene(deg_data_frame, 'ctrl_mean', 'ex_mean', 'ZNHIT1') # change to any gene in 'ZNHIT1'

3.7.4 Plot data in volcano plot and MA plot¶
# Define functions
def volcano_plot(deg_data_frame):
# input deg_data_frame from process_data
# returns volcano plot
fig, ax = plt.subplots()
volcano_plot = deg_data_frame.plot(x='log2(foldchange)', y='-log10(p-value)', c='p-value', kind='scatter', colormap='viridis', title = 'volcano plot', ax=ax)
def MA_plot(deg_data_frame):
# input deg_data_frame from process_data
# returns MA plot
fig, ax = plt.subplots()
MA_plot = deg_data_frame.plot(x='ctrl_mean', y='log2(foldchange)', c='p-value', kind='scatter', colormap='viridis', title='MA plot', ax=ax)
def save_deg_data(deg_data_frame, file_name, path):
# requires dataframe in the format generated from 'process_data'
# saves the file with the given name in the given location
final_path = os.path.join(path, f"{file_name}.csv")
deg_data_frame.to_csv(final_path)
# Volcano plot identifies changes in large data sets composed of replicate data.
volcano_plot(deg_data_frame)

# MA plot visualizes the differences between measurements taken in two samples, by transforming the data onto M (log ratio) and A (mean average) scales, then plotting these values.
MA_plot(deg_data_frame)

End of demo notebook on gene expresssion.
4. Analysis on structured metadata from the OpenAccess-CCLE project¶
4.1 Introduction to the dataset¶
- The project's data can be found here on the data model graph.
- The metadata we are interested in is in the node "lab_test".
- Metadata in the node "lab_test" include parameters associated with the result of a standardized, clinical laboratory test aimed at quantifying a particular molecule, analyte or biological marker in a biospecimen collected from a study subject.
# Import Gen3 SDK tools to the workspace
!pip install gen3
import gen3
from gen3.auth import Gen3Auth
from gen3.submission import Gen3Submission
# Useful commands to print and change current working directory
#os.getcwd() # print directory
#os.chdir('/home/jovyan') # change directory
# Authentication by calling the earlier downloaded credentials
endpoint = "https://gen3.datacommons.io/"
creds = "/home/jovyan/pd/gen_creds.json"
auth = Gen3Auth(endpoint, creds)
sub = Gen3Submission(endpoint, auth)
home_directory = '/home/jovyan/pd/dir_x' # the "dir_x" was created for demo purposes. Replace with a path if needed.
# Download the data associated to graph node using function "export_node"
lab_test = sub.export_node("OpenAccess", "CCLE", "lab_test", "tsv", home_directory +"/OA_CCLE_lab_test.tsv")
4.2 Read and clean (meta)dataset¶
lab_test_df = pd.read_csv('/home/jovyan/pd/dir_x/OA_CCLE_lab_test.tsv', sep ="\t")
lab_test_df.dropna(1) # remove columns that have "NaN"
- The column "sample_composition" shows the tissue type like "Central Nervous System" and the cell line like "G11".
# Creating a separate column for cell lines
lab_test_df['cell_line'] = lab_test_df['samples.submitter_id'].str.split('_', 1).str.get(0)
lab_test_df.columns
4.3 Plot a bar graph of categorical variable counts in a dataframe¶
# import libraries
from collections import Counter
from statistics import mean
import matplotlib.pyplot as plt
import numpy as np
import seaborn as sns
from sklearn.preprocessing import StandardScaler #for PCA
# Define function
def plot_categorical_property(property,df):
df = df[df[property].notnull()]
N = len(df)
categories, counts = zip(*Counter(df[property]).items())
y_pos = np.arange(len(categories))
plt.bar(y_pos, counts, align='center', alpha=0.5)
plt.xticks(y_pos, categories)
plt.ylabel('Counts')
plt.title(str('Counts by '+property+' (N = '+str(N)+')'))
plt.xticks(rotation=90, horizontalalignment='center')
#add N for each bar
plt.show()
# Plot a bar graph of categorical variable counts in a dataframe
plot_categorical_property("sample_composition", lab_test_df)

4.4 Plot a bar graph of categorical variable counts in order from largest to smallest¶
# Define function
def plot_categorical_property_by_order(property,df):
df = df[df[property].notnull()]
N = len(df)
categories, counts = zip(*df[property].value_counts().items()) # valuecounts orders it from largest to smallest
y_pos = np.arange(len(categories))
plt.bar(y_pos, counts, align='center', alpha=0.5)
plt.xticks(y_pos, categories)
plt.ylabel('Counts')
plt.title(str('Counts by '+property+' (N = '+str(N)+')'))
plt.xticks(rotation=90, horizontalalignment='center')
#add N for each bar
plt.show()
# Plot a bar graph of categorical variable counts in a dataframe
plot_categorical_property_by_order("sample_composition", lab_test_df)

4.5 Plot the probability PDF of a numeric property¶
# Define function
def plot_numeric_property(property,df,by_project=False):
df[property] = pd.to_numeric(df[property],errors='coerce') # This line changes object into float
df = df[df[property].notnull()]
data = list(df[property])
N = len(data)
fig = sns.distplot(data, hist=False, kde=True,
bins=int(180/5), color = 'darkblue',
kde_kws={'linewidth': 2})
plt.xlabel(property)
plt.ylabel("Probability")
plt.title("PDF for all projects "+property+' (N = '+str(N)+')') # You can comment this line out if you don't need title
plt.show(fig)
# Plots the probability of EC50
plot_numeric_property('EC50', lab_test_df)

# Plots the probability of the activity area
plot_numeric_property('activity_area', lab_test_df)

4.5 Scatter plot of numeric variables¶
def scatter_numeric_by_numeric(df, numeric_property_a, numeric_property_b):
df[numeric_property_a] = pd.to_numeric(df[numeric_property_a],errors='coerce') #BB: this line changes object into float
df = df[df[numeric_property_a].notnull()]
df[numeric_property_b] = pd.to_numeric(df[numeric_property_b],errors='coerce') #BB: this line changes object into float
df = df[df[numeric_property_b].notnull()]
data = list(df[numeric_property_a])
N = len(data)
plt.scatter(df[numeric_property_a], df[numeric_property_b])
plt.title(numeric_property_a + " vs " + numeric_property_b)
plt.xlabel(numeric_property_a)
plt.ylabel(numeric_property_b)
plt.show()
# Plots a scatter plot of two numeric variables, here EC50 vs IC50
scatter_numeric_by_numeric(lab_test_df, 'EC50', 'IC50')

# Plots a scatter plot of two numeric variables, here activity area vs maximum activity
scatter_numeric_by_numeric(lab_test_df, 'activity_area', 'max_activity')

4.6 Display the counts of each category in a categorical variable¶
# Define function
def property_counts_by_project(prop, df):
df = df[df[prop].notnull()]
categories = list(set(df[prop]))
projects = list(set(df['project_id']))
project_table = pd.DataFrame(columns=['Project','Total']+categories)
project_table
proj_counts = {}
for project in projects:
cat_counts = {}
cat_counts['Project'] = project
df1 = df.loc[df['project_id']==project]
total = 0
for category in categories:
cat_count = len(df1.loc[df1[prop]==category])
total+=cat_count
cat_counts[category] = cat_count
cat_counts['Total'] = total
index = len(project_table)
for key in list(cat_counts.keys()):
project_table.loc[index,key] = cat_counts[key]
project_table = project_table.sort_values(by='Total', ascending=False, na_position='first')
return project_table
property_counts_by_project("sample_composition", lab_test_df)
4.7 Display the counts of each category in a categorical variable in table form and sorted¶
# Define function
def property_counts_table(prop, df):
df = df[df[prop].notnull()]
counts = Counter(df[prop])
df1 = pd.DataFrame.from_dict(counts, orient='index').reset_index()
df1 = df1.rename(columns={'index':prop, 0:'count'}).sort_values(by='count', ascending=False)
#with pd.option_context('display.max_rows', None, 'display.max_columns', None):
display(df1)
display(df1.columns)
property_counts_table("sample_composition", lab_test_df)
4.8 Display the counts of each category in a pie chart and save image¶
# First, sort the amount of counts for a tissue, rename columns and show
sc_counts = lab_test_df.sample_composition.value_counts()
sc_counts = sc_counts.reset_index()
sc_counts = sc_counts.rename(columns={'index': 'sample_composition', 'sample_composition':'counts'})
sc_counts
# Second, return a pie chart of the counts for each category
data = sc_counts["counts"]
categories = sc_counts["sample_composition"]
fig1, ax1 = plt.subplots()
ax1.pie(data, labels=categories, autopct='%1.1f%%',
shadow=True, startangle=90)
ax1.axis('equal') # Equal aspect ratio ensures that pie is drawn as a circle.
plt.show()

- This pie chart shows too many entries. We will need to edit the amount of categories and we want to make changes to the color.
# Make a pie chart that shows only the categories with counts > 4000
top10 = sc_counts[sc_counts.counts > 4000].nlargest(10, 'counts')
data = top10['counts']
categories = top10["sample_composition"]
fig1, ax1 = plt.subplots()
# Changing the color of the pie
theme = plt.get_cmap('hsv')
ax1.set_prop_cycle("color", [theme(1. * i / len(top10))
for i in range(len(top10))])
ax1.pie(data, labels=categories, autopct='%1.1f%%',
shadow=True, startangle=90)
ax1.axis('equal') # Equal aspect ratio ensures that pie is drawn as a circle.
plt.show()

# Save the pie chart above
fig1.savefig('plot.png')
End of demo notebook. Please terminate your workspace session when finished.
See also other notebooks available here.